| Science | Diverses | Kinderseiten |
| |
Die etwas andere Homepage ... | ||
|
|
|
Topics:
Einfluß von Hitzeschock auf die ZellzyklusprogressionHitzeschock
ist ein Stressor, der praktisch alle zellulären Prozesse und Strukturen
beeinflußt. Aus den direkten Schädigungseffekten, wie Beeinträchtigung
der Membranintegrität, Proteindenaturierung und -aggregation sowie dem
Zerfall des Zytoskeletts resultieren zahlreiche primäre metabolische
Veränderungen. Die ATP-Gewinnung, die Makromolekülsynthese und der
intrazelluläre Transport werden gehemmt. In Abhängigkeit von der Schwere
der Schädigung brauchen Zellen nach dem Hitzeschock eine
unterschiedlich lange Zeitspanne, um den normalen Metabolismus
wiederherzustellen. Während dieser Erholungszeit treten weitere sekundäre
Effekte wie z.B. Zellzyklusverzögerungen und die Akkumulation von
Hitzeschockproteinen auf (Nover et al., 1984; Roti Roti et al., 1992;
Welch, 1992, Morimoto et al., 1996). In
zahlreichen Studien an primären Säugerzellkulturen sowie verschiedenen
Zellinien wurde gezeigt, daß nach Hitzeschock die G1-Progression gehemmt
und der S-Phaseeintritt verzögert ist (Van Dongen und Van Wijk, 1988;
Martin und Regan, 1991; Marui et al., 1991; Mackey et al., 1992; Wong et
al., 1993; Fuse et al., 1996; Nitta et al., 1997). Bei CHO-Zellen und
humanen Fibroblasten gibt es außerdem Hinweise, daß auch der G2/M-Übertritt
durch Hitzeschock verhindert wird (Hang und Fox, 1995; Sugano et al.,
1995). In besonders sensitiven, oft hematopoietischen Zellen dagegen
induziert bereits ein milder Hitzeschock Apoptose, während bei
resistenteren Zellinien hitzeinduzierter Zelltod durch Apoptose, Nekrose
oder den Verlust der Klonogenität erst bei Temperaturen ab 44°C oder
langanhaltendem (chronischem) Hitzeschock auftritt (Dewey et al., 1990;
Mackey et al., 1992; Roti Roti et al., 1992; Yonezawa et al., 1996; Van
der Waal et al., 1997). Zu
den offensichtlichsten und bestuntersuchtesten sekundären Wirkungen eines
Hitzeschocks gehören die Hemmung der allgemeinen Proteinsynthese und die
Akkumulation von Hitzeschockproteinen (HSP) während der Erholungszeit.
Nach ihrer Größe werden Ubiquitin, kleine HSP, HSP60-, HSP70- und
HSP90-Familien sowie HSP110 unterschieden (Nover et al., 1984; Welch,
1992; Ovelgonne und Van Wijk, 1995; Morimoto et al., 1996). Der Terminus
Hitzeschockproteine ist hierbei in zweifacher Weise irreführend.
Einerseits werden diese Proteine auch durch andere Stressoren wie Aminosäureanaloge,
Schwermetalle, Arsenit, Ethanol und oxidativen Streß induziert (Welch,
1992; Neuhaus-Steinmetz et al. 1994; Gosslau, 1998). Andererseits wird die
Mehrzahl der HSP auch unter physiologischen Bedingungen exprimiert. Nur im
Fall der HSP70-Familie wird dieser Unterschied von einigen Autoren in der
Namensgebung kenntlich gemacht. Die konstitutive, zytosolische Form wird
als heat shock cognate (HSC70)
bezeichnet, während die am stärksten induzierbare Form HSP70 genannt
wird. Im Zusammenhang mit den artspezifischen Molekulargewichten dieser
beiden Proteine werden außerdem verschiedene Namen wie HSC73 und HSP73 für
HSC70 bzw. HSP72 und HSP68 für HSP70 synonym verwendet (Subjeck und Shyy,
1986). Mittlerweile
ist allgemein anerkannt, daß Hitzeschockproteine auch unter
physiologischen Bedingungen essentielle zelluläre Funktionen haben.
Ubiquitin, ein kleines, globuläres 8,5 kDa Protein ist unter
konstitutiven Bedingungen wie bereits beschrieben durch seine
Markierungsfunktion
für den 26 S Proteasom-abhängigen Abbau an der Zellzyklusregulation
beteiligt. Weitere Funktionen der Ubiquitinierung sind u.a. die Regulation
der Chromatin- und wahrscheinlich der Zytoskelettstruktur. Nach
Hitzeschock wird Ubiquitin verstärkt exprimiert und trägt zum Abbau
irreversibel denaturierter Proteine bei (Parsell und Lindquist, 1993; Kühl,
1995; Hochstrasser, 1996; Pagano, 1997; Pickard, 1997). Die
HSP60-, HSP70- und HSP90-Familien dagegen wirken als sogenannte Chaperone
(Becker und Craig, 1994; Hartl et al., 1994; Höhfeld, 1998; Ranson et
al., 1998). Hierunter versteht man im Fall der HSP70-Familie, daß sie
neusynthetisierte, ungefaltete Proteine stabilisieren, die Aggregation
verhindern sowie ihre Faltung und Kompartimenttranslokation unterstützen
(Beck und De Maio, 1994; Hartl et al., 1994; Beissinger und Buchner,
1998). Nach Hitzeschock wird HSC70 verstärkt exprimiert und HSP70
induziert. Beide Formen translozieren außerdem für einige Stunden verstärkt
in den Kern und die Nukleoli (Kühl, 1995; Ovelgonne und Van Wijk, 1995;
Zeise, 1996; Thayer und Mirkes, 1997; Gosslau, 1998; Helmbrecht, 1998).
Die
Bindung von HSC und HSP70 an Zellzyklus-relevante Proteine wie p53, c-Myc,
pRb und das große T-Antigen des simian
virus 40 (SV40) (Henriksson et al., 1992; Agoff et al., 1993; Okuno et
al., 1993; Inoue et al., 1995) sowie ihre S-Phase-spezifische Expression
und Kerntranslokation (Sainis et al., 1994; Konishi et al., 1995; Taira et
al., 1997; Zeise et al., 1998) waren und sind Kriterien, die Anlaß geben,
eine Rolle dieser HSP in der Zellzykluskontrolle zu vermuten (Pechan,
1991; Sato und Torigoe, 1998). In Übereinstimmung hiermit erhöht die künstliche
Überexpression von humanem HSP70 das Potential der murinen WEHI
Fibrosarkomzellinie nach Injektion in Mäuse die Tumorbildung auszulösen
(Jäättelä, 1995) und verhindert einen Doxorubicin-induzierten G2-Arrest
(Karlseder et al., 1996). Die Applikation von hsp70-antisense
Oligonukleotiden dagegen verhindert den G1/S-Übergang in humanen
Tumorzellen (Wei et al., 1995). Trotz
dieser deutlichen Hinweise wird die Funktion der Hitzeschockproteine im
Zellzyklus immer noch kontrovers diskutiert. Einige Autoren postulieren,
daß nur das induzierbare HSP70 eine Zellzyklusfunktion erfüllt und diese
nur nach Hitzeschock relevant wird (Hang und Fox, 1996; He und Fox, 1996).
Besonders kontrovers wird die Rolle der zytosolischen HSP70-Formen auch im
Zusammenhang mit der Induktion von erworbener Thermotoleranz
diskutiert (Borelli et al., 1996; Akagawa et al., 1998). Hierunter
versteht man, daß ein milder Hitzeschock Zellen gegenüber einem zweiten,
starken Streß resistenter macht. Dieses Phänomen wurde für alle oben
genannten primären und sekundären Effekte von Hitzeschock beschrieben (Pelham,
1984; Mizzen und Welch, 1988; Parsell und Lindquist, 1993; Beck und De
Maio, 1994; Kim et al., 1995; Ovelgonne und Van Wijk, 1995; Wong et al.,
1995). In Bezug auf den Zellzyklus versteht man unter Thermotoleranz, daß
die Anzahl an Zellen, die einen letalen Hitzeschock überlebt deutlich erhöht
wird (Roti Roti et al., 1992; Borelli et al., 1996; Akagawa et al., 1998). Während
einige Autoren den thermotoleranten Status nur auf die verstärkte
Expression und Kerntranslokation von HSC und HSP70 zurückführen, sehen
Andere auch die kleinen HSP als Vermittler von Thermotoleranz an (Mizzen
und Welch, 1988; Laszlo et al., 1993; Thayer und Mirkes, 1997). In einigen
Fällen deutet die Thermotoleranzausbildung bei gleichzeitiger Hemmung der
HSP-Synthese auch auf vollkommen unabhängige Mechanismen hin (Borelli et
al., 1996).
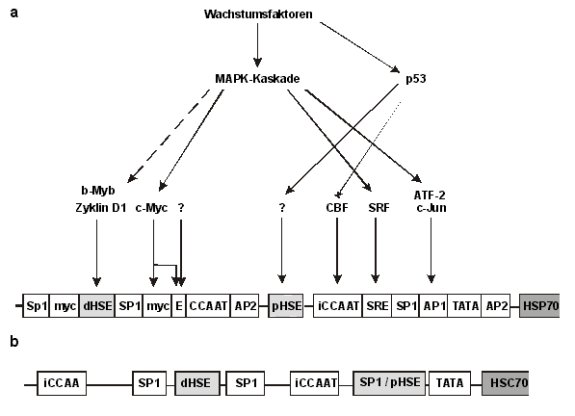
Regulation der konstitutiven Expression von HitzeschockproteinenObwohl der Promotor des HSC70-Gens der Ratte bereits 1987 von Sorger und Pelham veröffentlicht wurde, gibt es bisher keine Untersuchungen die belegen, wie die zellzyklusabhängige Expression von HSC70 zustande kommt (Abb. 4.3-1b). Die G1-spezifische Induktion könnte in Analogie zur konstitutiven hsp72-Aktivierung in humanen Zellinien über das distale und proximale HSE verlaufen (Abb. 4.3-1a). Kamano und Klempnauer (1997) konnten zeigen, daß Zyklin D1 und b-Myb das distale HSE des hsp72-Promotors transaktivieren. Die Aktivierung ist unabhängig von der DNA-Bindungsdomäne in b-Myb und der Phosphorylierung von pRb durch die Zyklin D1-abhängige Cdk4. Beide HSE sowie die CCAAT-Boxen werden indirekt sowohl von Wildtyp als auch mutiertem p53 reguliert. Die Transaktivierung der HSE verläuft indirekt über einen nicht bekannten Mechanismus, während eine Reprimierung der CCAAT-stimulierten Transkription über die Bindung und Inaktivierung von CBF durch p53 vermittelt wird (Agoff et al., 1993; Tsutsumi-Iishi et al., 1995).
Abb.: Promotor des humanen HSP72-Gens und des HSC70-Gens der Ratte a: Konstitutive
Signalwege der hsp72-Induktion
in humanen Zellen. Bisher sind drei Hauptwege der hsp72-Induktion in ungestreßten Zellen bekannt. Die MAPK-Kaskade
stimuliert die HSP72-Expression indirekt durch Transaktivierung des SRE,
der AP-1/ATF-2-Bindungsstelle und zwei c-Myc-Bindungsstellen (Hunt und
Morimoto, 1985; Wu et al., 1987; Tsutsumi-Iishi et al., 1995). Das distale
und proximale HSE werden durch b-Myb und Zyklin D1 bzw. p53 stimuliert (Tsutsumi-Iishi
et al., 1995;
Kamano
und Klempnauer, 1997). Der dritte Weg verläuft über ein enhancer-Element, das in Reportergenkonstrukten G1-
phasenspezifische Expressionsmuster induziert. c-Myc wurde als
potentieller Transaktivator identifiziert, da es zusammen mit anderen,
nicht identifizierten Proteinen an das Verstärkerelement bindet (Taira et
al., 1997). b: Promotor des
HSC70-Gens der Ratte (Sorger und Pelham, 1987). Bisher gibt es keine
Studien, die belegen, welche Promotorelemente für die konstitutive
Expression verantwortlich sind. In Analogie zur Regulation des humanen hsp72-Promotors
könnte die konstitutive Transaktivierung über die HSE verlaufen. AP1:
Bindungsstelle für AP-1, c-Jun-Homodimere und ATF-2. Die Sequenz enthält
auch ein CRE (cAMP-responsives Element). AP-2:
AP-2-Bindungsstelle. CCAAT:
CCAAT-Box. dHSE: distales
Hitzeschockelement. E: enhancer
element. iCCAAT:
invertierte CCAAT-Box. myc:
Bindungsstelle für c-Myc. pHSE:
proximales HSE. Das pHSE im HSC70-Gen überlappt mit einer
SP1-Bindungsstelle. SP1: SP1-Bindungsstelle. SRE:
serum response element. Bindungsstelle
des SRF. SRF: serum response factor. TATA: TATA-Box.
Streßinduzierte Expression von HitzeschockproteinenDie
selektive Transkription der Hitzeschockproteingene verläuft nach
Hitzeschock über die Aktivierung und Bindung des HSF1 und HSF3 an
Hitzeschockelemente in den entsprechenden Promotoren. Der HSF1 wird nach
Hitzeschock innerhalb von Minuten aktiviert und scheint für die schnelle
Induktion der Hitzeschockproteine verantwortlich zu sein. Der HSF3 dagegen
wird erst bei höheren Temperaturen aktiviert und zeigt eine verzögerte
Bindungskinetik. Seine genaue Rolle bei der Induktion von
Hitzeschockproteinen ist bisher nicht geklärt (Sarge et al., 1993;
Morimoto et al., 1996; Tanabe et al., 1998). In ungestreßten Zellen
unterliegt der HSF1 zwei generellen Repressionsmechanismen (Abb.). Ein
Multichaperonkomplex, der u.a. konstitutive HSP70-Formen und HSP90 enthält,
bindet und inhibiert HSF1-Monomere. Hierdurch unterliegt die
HSP-Expression einer negativen Rückkopplung (Rabindran et al., 1993;
Baler et al., 1996; Zou et al., 1998). Die MAPK, die
Glykogensynthase-Kinase 3-b
sowie die PkC-a
und -z
phosphorylieren den HSF1 inhibitorisch an verschiedenen Serinresten (Kline
und Morimoto, 1997; Chu et al., 1998).
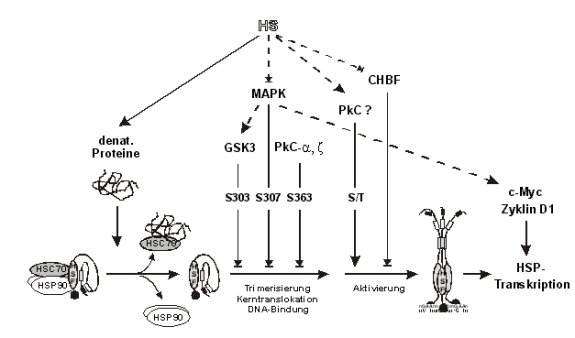
Abb.: Regulation der streßinduzierten Expression von
Hitzeschockproteinen Vereinfachte Darstellung der HSF1-Regulation
und Expression von Hitzeschockproteinen. In ungestreßten Zellen wird die
Transaktivierungskapazität des HSF1 über zwei Mechanismen blockiert.
Konstitutive Chaperonkomplexe (u.a. HSC70 und HSP90) binden den HSF1 und
halten ihn in einer monomeren, inaktiven Form (Rabindran et al., 1993;
Baler et al., 1996; Zou et al., 1998). Die Trimerisierungs- und
DNA-Bindungsfähigkeit der HSF1-Monomere ist außerdem durch mindestens
drei inhibitorische Phosphorylierungen gehemmt. Die initiale
Phosphorylierung des Serin 307 durch die MAPK ermöglicht die
GSK3-katalysierte Phosphorylierung von S303. Die GSK-3 wird außerdem
indirekt durch die MAPK aktiviert. Die PkC ist wahrscheinlich für die
Phosphorylierung von S363 verantwortlich (Kline und Morimoto, 1997; Chu et
al., 1998). Die Expression von Hitzeschockproteinen verläuft über
konstitutive Transaktivatoren wie z.B. c-Myc oder Zyklin D1 (vgl. auch
4.3-1). Nach Hitzeschock wird der HSF1 aktiviert, und Teile der
konstitutiven Induktionskaskade werden abgeschaltet. Denaturierte Proteine
verdrängen den HSF1 aus der Bindung an den Chaperonkomplex (Kim et al.,
1995; Baler et al., 1996; Zou et al., 1998). Die freien Monomere werden
wahrscheinlich nicht mehr inaktivierend phosphoryliert, weil der
Hitzeschock u.a. MAPK-spezifische Phosphatasen (MKP) induziert (Ishibashi
et al., 1994; Nebreda, 1994 ), die MAPK inaktiviert und somit zumindest
die S303- und S307-Phosphorylierung unterbindet. Der HSF1 kann
trimerisieren, in den Kern translozieren und an Hitzeschockelemente in den
HSP-Promotoren binden (Baler et al., 1993; Sarge et al., 1993). Zur
Transaktivierung muß der HSF1 jedoch zusätzlich aktivierend an Serin-
und Threoninresten phosphoryliert werden (Cotto et al., 1996; Xia und
Voellmy, 1997). Die PkC wird nach Hitzeschock aktiviert, katalysiert diese
Modifikationen in vitro und könnte
somit für diese Aktivierung verantwortlich sein (Ding et al., 1997;
Ohnishi et al., 1998). CHBF: constitutive HSE-binding factor. GSK3: Glykogensynthase-Kinase 3. MAPK:
Mitogen-aktivierte Proteinkinasen Erk1 und Erk2. PkC-a, z:
Proteinkinase C a- oder z-Isoform.
®: positiver
Einfluß. ®|: negativer Einfluß. Pfeile mit durchgezogenen oder
unterbrochenen Linien kennzeichnen direkte bzw. indirekte Wirkungen.
Nach
Hitzeschock wird der HSF1 posttranskriptional aktiviert; eine
hitzeinduzierte Steigerung der Expression wurde bisher für Säugerzellen
nicht beschrieben (Feige und Polla, 1994; Wu et al., 1995). Die
Aktivierung verläuft über die Entlassung aus den Multichaperonkomplexen
(Morimoto et al., 1996; Zou et al., 1998), die Trimerisierung,
Kerntranslokation und HSE-Bindung (Baler et al., 1993; Sarge et al., 1993;
Baler et al., 1996) sowie die Hyperphosphorylierung an mindestens 4
aktivierenden Serin- und Threoninresten (Cotto et al., 1996; Xia und
Voellmy, 1997; Ding et al., 1997; Ohnishi et al., 1998). Neben diesen
direkten Effekten auf den HSF1 wird die Transkription der HSP-Gene zusätzlich
durch die Inhibierung des konstitutiv HSE-bindenden Faktors (CHBF) ermöglicht.
Die HSE-Bindung des HSF1 und des CHBF ist kompetitiv, und erst die
Inaktivierung des CHBF durch Hitzeschock ermöglicht die Induktion der
Hitzeschockproteine (Liu et al., 1994, 1995). Dies könnte eine Erklärung
dafür liefern, daß bei milden Temperaturen trotz HSF1-Aktivierung keine
Transkription von Streßgenen stattfindet (Liu et al., 1994, 1995; Wu,
1995; Zuo et al., 1995). Soweit die Vorgänge bisher aufgeklärt sind,
verläuft die Inaktivierung des HSF1 nach ausreichender Akkumulation von
Hitzeschockproteinen über die Umkehrung dieser Prozesse (Wu, 1995; He et
al., 1998). Phosphatasen, die die aktivierenden oder inhibitorischen Phosphorylierungen des HSF1 entfernen sind bisher nicht identifiziert. Daher ist nicht klar, ob die MAPK zur HSF1-Aktivierung abgeschaltet werden muß, oder ob sich das Verhältnis zwischen Kinase- und Phosphataseaktivität verändert. Die Ergebnisse in Bezug auf die MAPK-Aktivität nach Hitzeschock sind widersprüchlich. In zahlreichen Studien wurde eine kurzfristige Aktivierung der MAPK nach Hitzeschock beschrieben (Kyriakis et al., 1995; Kline und Morimoto, 1997; Lewis et al., 1998). Andererseits konnte nach Hitzeschock und anderen Stressoren die transkriptionale Induktion von MAPK-spezifischen Phosphatasen (MKP), sogenannten dual-spezifischen Serin/Threonin und Tyrosin-Phosphatasen, gezeigt werden, die die MAPK inaktivieren (Sun et al., 1993; Ishibashi et al., 1994, Nebreda, 1994; Lewis et al., 1998).
Einfluß von Hitzeschock auf Regulatoren der Zellzyklusprogression
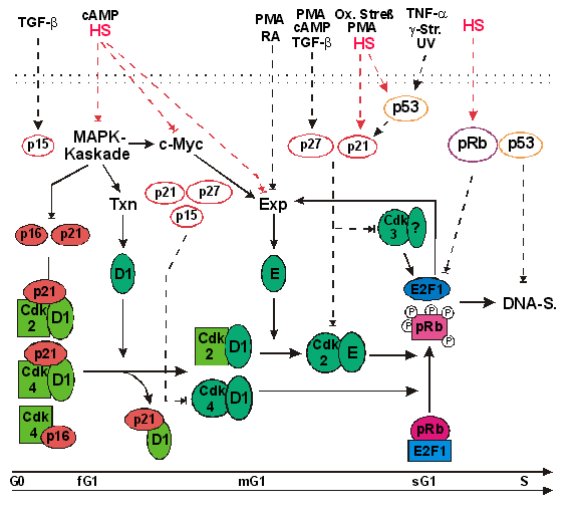
Abb.:
Modell der streßinduzierten Verzögerung des G1/S-Übergangs Zu jedem Zeitpunkt in G1 können Stressoren,
Zytokine oder Proteinkinaseaktivatoren die Reaktionsfolge, die zur
pRb-Phosphorylierung und E2F-1-Freisetzung führt, verzögern oder vollständig
inhibieren. Die meisten Stressoren induzieren einen oder mehrere
Cdk-Inhibitoren. Die p16-ink4-Familie hemmt nur die Cdk4 und 6, während
p21 und p27 alle Cdk-Komplexe hemmen (Kato et al., 1994; Serrano, 1997).
p21-cip1/waf1 wird nach Hitzeschock über einen p53-abhängigen und einen
p53-unabhängigen Weg induziert (Fuse et al., 1996; Ohnishi et al., 1996;
Nitta et al., 1997). Neben diesem Hauptmechanismus wird die
Wachstumshemmung je nach Induktor zusätzlich durch weitere Effekte verstärkt.
Die Hemmung der MAPK-Kaskade durch Hitzeschock und cAMP verhindert
wahrscheinlich indirekt die Zyklin D-Akkumulation und den Abbau von p16
und p21 (L´Allemain et al., 1997; Lewis et al., 1998; Roussel, 1998). Die
Hemmung der c-Myc-Expression könnte ebenfalls über die MAPK oder über
einen der anderen c-Myc-Induktionswege verlaufen (Bukh et al., 1990;
Roussel, 1998). Hitzeschock, cAMP, RA und PMA verhindern außerdem die
Zyklin E-Expression und blockieren so die Vollendung der
pRb-Phosphorylierung (vgl. 3.6.2; Gagelin et al., 1994; Khandijan et al.,
1995; Livneh und Fishman, 1997; Lee et al., 1998). Dies verhindert die
positive Rückkopplung zwischen E2F-1-Freisetzung und der Zyklin
E-Akkumulation, die die dramatische Zunahme der Cdk2/E- und E2F-1-Aktivität
sowie den Eintritt in die S-Phase auslöst (Bartek et al., 1997; Bateman,
1998; Johnson und Schneider-Broussard, 1998). g-Str.: g-Strahlung.
DNA-S.: DNA-Synthese. E2F1:
E2F-1 gebunden an DP1 oder DP2. Exp:
Expression (Transkription und mRNA-Stabilität). f,
m, sG1: frühe, mittlere und späte G1-Phase. PMA:
Phorbolmyrestylacetat (PkC-Aktivator). RA: retinoic
acid. TGF-b: transforming growth factor b. TNF-a: Tumornekrosefaktor a. Txn: Transkription. Kreise:
kennzeichnen aktive Cdk-Formen. Quadrate:
Kennzeichnen inaktive Cdk-Formen. Weiße
Ovale kennzeichnen die streßinduzierte Expression dieser Regulatoren.
Pfeile mit durchgezogenen oder unterbrochenen Linien stellen
konstitutive bzw. streßinduzierte Wirkungen dar.
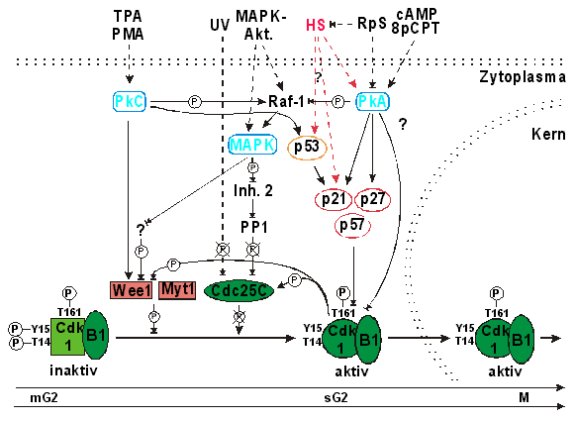
Abb.:
Modell der streßinduzierten Blockade des G2/M-Übergangs Zielpunkt aller bekannten G2-arretierenden Signale
ist die Aktivierung des MPF (Cdk1/B1). Die genauen Mechanismen, die zur
Hemmung des MPF führen sind jedoch nur selten bekannt. UV-Strahlung, MAPK-
und PkC-Aktivatoren hemmen die Cdk1-aktivierende Phosphatase Cdc25C und/oder
aktivieren die Cdk1-inhibierende Kinase Wee1 indirekt (Livneh und Fishman,
1997; Poon et al., 1997; Walter et al., 1997; Lewis et al., 1998). Der
Weg, über den die PkA die Hemmung des MPF bewirkt, ist vollkommen unklar.
Ein Mechanismus könnte die Induktion der Cdk-Inhibitoren p21 und p27 sein
(Lamb et al., 1991; Kato et al., 1994; Niculescu et al., 1998). Es ist
jedoch nicht auszuschließen, daß neben den dargestellten Wirkungen auch
direkte Effekte auf die Cdk1 oder die regulierenden Kinasen und Phosphatasen
eine Rolle spielen. Der Mechanismus der G2-Arretierung nach Hitzeschock
ist ebenfalls unbekannt. In Analogie zur G1-Arretierung kommt als Zielpunkt
der Cdk-Inhibitor p21 in Frage. In C6-Zellen scheint die G2-Arretierung
außerdem von der Aktivierung der PkA abhängig zu sein. Die Hemmung der
PkA ist unter konstitutiven Bedingungen für den G2/M-Übergang notwendig
(Beebe, 1994) und nach Hitzeschock in C6-Zellen ausreichend, um die G2-Blockade
aufzuheben. In allen bisher untersuchten Zelltypen erfolgt die streßinduzierte
G2-Arretierung vor der Kerntranslokation des MPF (King et al., 1994; Rieder
und Khodjakov, 1997). 8pCPT: 8-pCPT-cAMP, PkA-Aktivator. B1:
Zyklin B1. Cdc25C: Phosphatase. Inh. 2: Inhibitor
2. Myt1: Kinase. PMA:
Phorbolmyrestylacetat, PkC-Aktivator. PP1:
Proteinphosphatase 1. RpS:
Rp-8-CPT-cAMPS, PkA-Inhibitor. TPA: 12-O-Tetradecanoylphorbol-13-acetat, PkC-Aktivator.
Wee1:
Kinase. Pfeile mit durchgezogenen
oder unterbrochenen Linien stellen konstitutive bzw. streßinduzierte Effekte
dar. |
Copyright Information Portions of this work may be individually downloaded, copied, and cited for the personal and educational purposes of individuals and organizations in their work, provided that proper attribution and context are given. Commercial reproduction or multiple distribution of any kind is prohibited. All rights reserved. Reproduction in whole or in part in any form or medium without express written permission of the author is prohibited (kinetophil@yahoo.com). |